Larrayoz MJ and Calasanz MJ, laboratory of Genetics Hematologic and Co-Scientific Director of CIMA LAB Diagnostics
Precision medicine in onco-hematology: implementation of massive sequencing in the integrated diagnosis of hematological malignancies.
The diagnosis of hematologic malignancies has changed substantially in recent decades, moving from morphologic assessment as the sole criterion, to the integration of clinical, morphologic, immunophenotypic, cytogenetic and molecular findings, which are the basis of the World Health Organization (WHO) classification. The new genetic data have transformed our understanding of the pathophysiology of hematologic malignancies, implicating new biological pathways and providing a new tool to deconstruct the complexity of these pathologies. In recent years there has been an explosion of new molecular data thanks to the use of massive sequencing techniques (Next-Generation Sequencing, NGS), both whole genomes (WGS), exomes (WES), and panels directed to the analysis of certain genes (Fernandez-Mercado, 2013; Dubois, 2016). The recentSpecial Series of the Journal of Clinical Oncology comes to reinforce this vision of the importance of genomic medicine in hematologic oncology (Ebert, 2017).
We are once again witnessing a crucial moment of paradigm shift in onco-hematological diagnosis, with the incorporation of the concept of precision medicine (also called personalized medicine). We already experienced a similar status (profound change) three decades ago with the incorporation of cytogenetic analysis into our laboratories. Today karyotyping/FISH are essential for diagnosis, classification, prognostic stratification and therapeutic guidance in hematologic malignancies. At this moment, the new challenge ahead of us is the incorporation of massive sequencing to the integrated diagnosis of hematological malignancies.
It is true that we have been routinely performing for years, in addition to conventional cytogenetic techniques (karyotyping and FISH), some molecular tests that are necessary for both diagnosis and prognostic stratification in lymphomas, multiple myeloma, chronic lymphocytic leukemia and other lymphoid neoplasms (Campo, 2011; Swerdlow, 2016; Rosenquist, 2016). The same is true in myeloid pathology, with the analysis of CEBPA, NPM1 and FLT3 in acute myeloid leukemia (AML); although this list of molecular tests could be lengthened shortly, as recent data point to mutations in other genes that could also have clinical value in AML (Papaemmanuil, 2016). In the case of myelodysplastic syndromes (MDS), it has also been suggested that the IPSS-R (Revised International Prognostic Scoring System, Greenberg, 2012) could be improved by incorporating molecular data (Nazha, 2016). As we say, these molecular markers are already being analyzed for diagnostic purposes by classical molecular biology techniques (Sanger sequencing, ARMS-PCR, ASO-PCR or RT-qPCR), integrated with the data cytomorphology, cytometry and cytogenetics.
However, the large number of new molecular data available, together with the accessibility of NGS, has opened a new door to be able to implement the new markers into the protocols of molecular pathology diagnostic laboratories, so that the genetic diagnosis we currently perform can become genomic. In fact, several commercial companies that include massive sequencing in their product portfolio are expanding their offering of NGS panels designed to analyze specific pathologies. These targeted panels are capable of simultaneously analyzing regions of 20-60 genes, or even entire genes, selected based on relevance to the pathology to be studied. In economic terms, the large amount of genomic information produced by massive panel sequencing technology would be more expensive and more laborious to obtain by Sanger sequencing. In addition, the simultaneous analysis of such a large number of genes issue drastically reduces (even by half) the turnaround times, compared to the sequential gene analysis schemes often applied in molecular laboratories (Figure 1). In addition to the undoubted advantages in terms of time and cost, this technology makes it possible to detect emerging clones that can help in the monitoring and prognosis of the disease, and thus detect a possible change of course in its history, perhaps associated with resistance to treatment or other factors. Finally, the availability of such detailed molecular information will allow the rapid identification of patients who may benefit from clinical trials that include these molecular aberrations in their inclusion criteria.
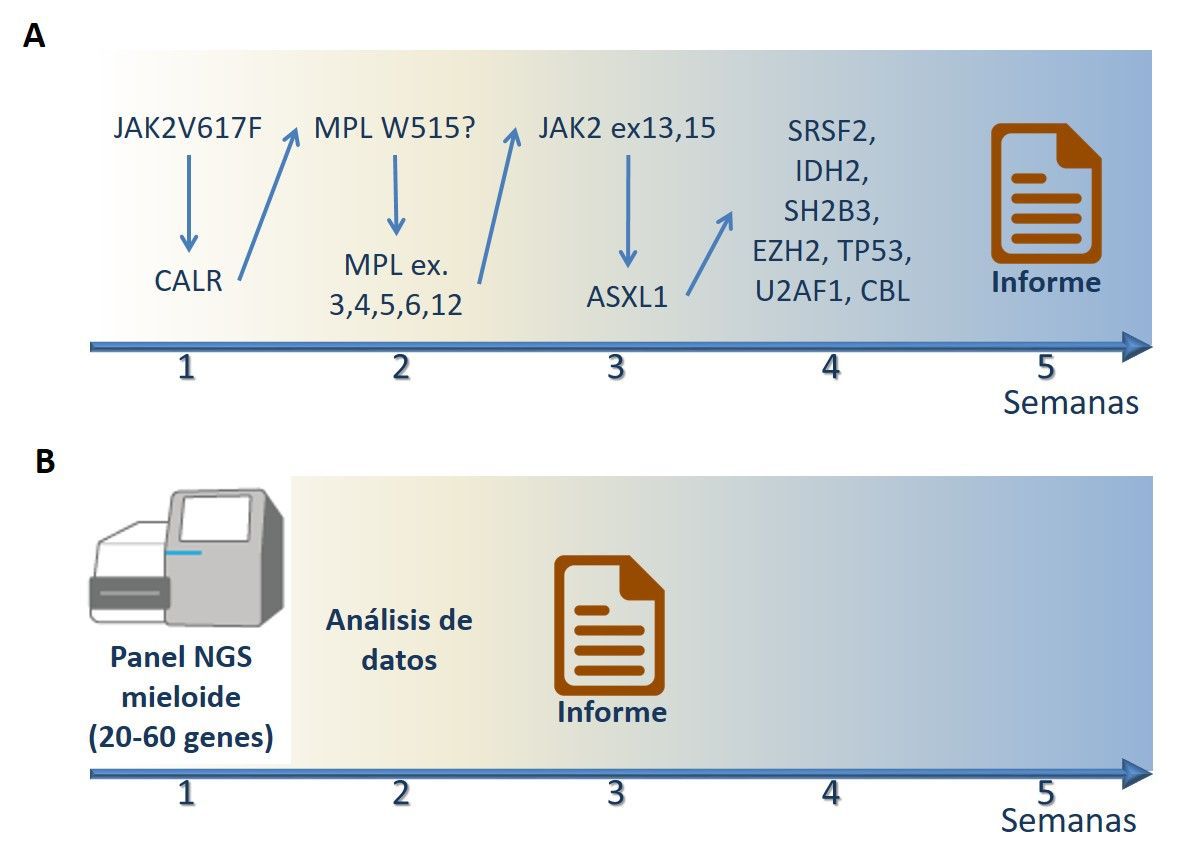 |
Figure 1. Flows from work to the issuance of the molecular pathology report when analyzing a sample suspected of idiopathic myelofibrosis. (A) outline of sequential genetic analysis by classical molecular biology techniques (ARMS-PCR and Sanger sequencing). Each negative test requires a new molecular test. The whole process can take up to 5 weeks until the issuance of report. (B) outline of genomic analysis by NGS. All relevant genes associated with the pathology are sequenced at the same time. After computer processing of the sequencing data , the report is issued. The whole process can take up to 15-20 days. |
However, there are some problems with the incorporation of massive sequencing into diagnostics that are delaying the standardization of its use. On the one hand, the complexity of the data generated by the sequencers requires staff highly qualified in computational biology, which for the moment is only possible in diagnostic laboratories with a strong commitment to research. On the other hand, the scarcity of international guidelines for onco-hematological diagnosis means that laboratories that have begun to use this technique have to make a great effort to take an overall view of the results they generate, to detect recurrent errors in sequencing, to discern the possible implications of incidental findings, to develop lines of data analysis adapted to each panel, and to prepare reports that are of real clinical utility. Finally, from a technical standpoint, we have experience that not all genes are analyzed with equal accuracy by this technique. For example, long insertions and deletions often escape the standard computational analyses applied on the sequencing data ; similarly, genomic regions that are encoded in GC-rich sites are difficult to capture in the initial steps of preparing the sample prior to sequencing. For these reasons, we foresee a near future in which NGS will probably not supplant molecular techniques already installed in laboratories, but rather coexist with them, at least for a few years.
In addition, massive sequencing platforms generate such a large number of data, that in addition to variants known to be associated with the disease, others of uncertain significance are identified. Laboratories that incorporate NGS into the integrated diagnosis of hematological malignancies will benefit from coordinated work with other laboratories using the same techniques, as together it will be easier to build the critical mass of mass sequencing data to interpret the significance of genetic variants of uncertain significance (VUS). For the time being, the resource is to go to instructions of data of variants with data of population frequency to infer whether the detected VUS can be related to the pathology or not.
On the other hand, it is possible that this technology will not be extended indiscriminately to all diagnostic laboratories that currently perform molecular testing, but will be restricted to a few laboratories at reference letter throughout the country. This could be due not only to the high initial investment in equipment and the need for a highly qualified staff , but also to the fact that in order to guarantee good response times and affordable prices there must be a sufficient flow of samples.
We are witnessing a historic moment, a new turning point in hematological diagnostics. NGS is no longer just a technology of the large groups of research, but is being implemented in routine diagnosis. We are aware that this will require intense partnership among specialists in our country on issues such as the standardization of techniques (endorsed by the SEHH), the training of professionals or the accreditation of procedures. Clinicians, pathologists, bioinformaticians and geneticists must work more closely than ever to ensure that this new technology of enormous potential is implemented as a new diagnostic technique and is of real clinical benefit.
Acknowledgements
The authors would like to thank the entire team of CIMA LAB Diagnostics. MFM is also grateful for funding from the association Española Contra el Cáncer (AECC), the Diputación Foral de Guipuzcoa (DFG15/011) and the high school de Salud Carlos III (PI16/00159).
References
Campo E et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011 May 12;117(19):5019-32. doi:http://dx.doi.org/10.1182/blood-2011-01-293050
Dubois S et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res. 2016 Jun 15;22(12):2919-28. doi: http://dx.doi.org/10.1158/1078-0432.CCR-15-2305
Ebert B and Friedberg JW. Introduction to Genomics in Hematologic Malignancy. Journal of Clinical Oncology. 2017 [in press] doi: http://dx.doi.org/10.1200/JCO.2017.72.2827
Fernandez-Mercado M et al. Targeted re-sequencing analysis of 25 genes commonly mutated in myeloid disorders in del(5q) myelodysplastic syndromes. Haematologica. 2013 Dec;98(12):1856-64. doi: http://dx.doi.org/10.3324/haematol.2013.086686
Greenberg PL et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012 Sep 20;120(12):2454-65 doi: http://dx.doi.org/10.1182/blood-2012-03-420489
Nazha A et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia. 2016 Nov;30(11):2214-2220. doi: http://dx.doi.org/10.1038/leu.2016.138
Papaemmanuil E et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016 Jun 9;374(23):2209-2. doi: http://dx.doi.org/10.1056/NEJMoa1516192
Rosenquist R et al. Clinical impact of recurrently mutated genes on lymphoma diagnostics: state-of-the-art and beyond. Haematologica. 2016 Sep;101(9):1002-9. doi:http://dx.doi.org/10.3324/haematol.2015.134510
Swerdlow SH et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016 May 19;127(20):2375-90. doi: http://dx.doi.org/10.1182/blood-2016-01-643569
*DOWNLOAD THE COMPLETE article IN PDF FORMAT